A Complete Medical Guide and Prevention For Heart Diseases Volume XIX; Marfan Syndrome
Nonfiction, Health & Well Being, Health, Ailments & Diseases, Heart, Health Care Issues
| Author: | Medical Professionals | ISBN: | 1230000037162 |
| Publisher: | MedHealth | Publication: | December 6, 2012 |
| Imprint: | Language: | English |
| Author: | Medical Professionals |
| ISBN: | 1230000037162 |
| Publisher: | MedHealth |
| Publication: | December 6, 2012 |
| Imprint: | |
| Language: | English |
Marfan syndrome (also called Marfan's syndrome) is a genetic disorder of the connective tissue. People with Marfan tend to be unusually tall, with long limbs and long, thin fingers.
The syndrome is inherited as a dominant trait, carried by the gene FBN1, which encodes the connective protein fibrillin-1. People have a pair of FBN1 genes. Because it is dominant, people who have inherited one affected FBN1 gene from either parent will have Marfan syndrome.
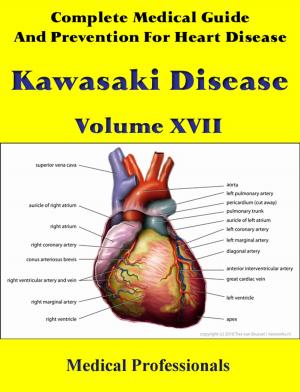
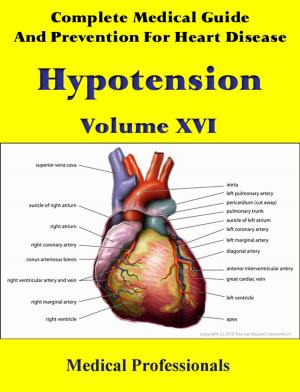
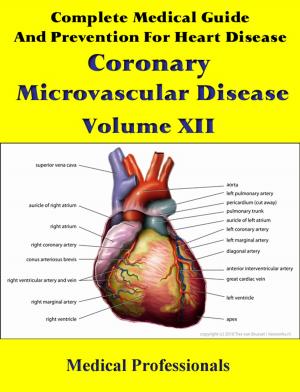
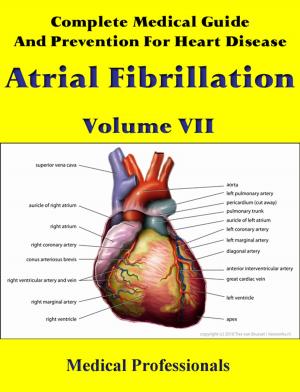
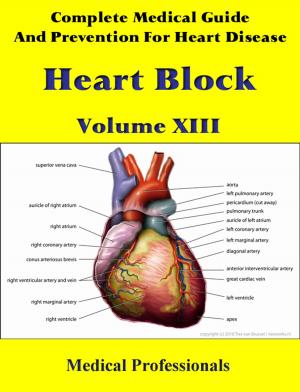
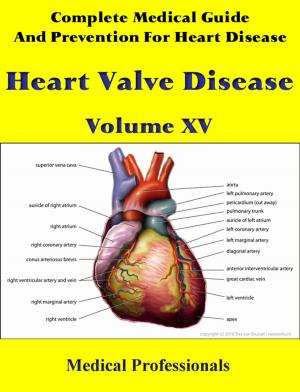
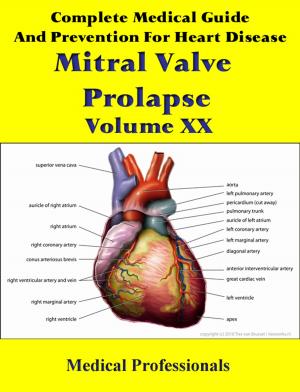
Marfan syndrome has a range of expressions, from mild to severe. The most serious complications are defects of the heart valves and aorta. It may also affect the lungs, the eyes, the dural sac surrounding the spinal cord, the skeleton and the hard palate.
In addition to being a connective protein that forms the structural support for tissues outside the cell, the normal fibrillin-1 protein binds to another protein, transforming growth factor beta (TGF-β). TGF-β has deleterious effects on vascular smooth muscle development and the integrity of the extracellular matrix. Researchers now believe, secondary to mutated fibrillin, excessive TGF-β at the lungs, heart valves, and aorta weakens the tissues and causes the features of Marfan syndrome. Since angiotensin II receptor antagonists (ARBs) also reduce TGF-β, ARBs (losartan, etc.) have been tested in a small sample of young, severely affected Marfan syndrome patients. In some patients, the growth of the aorta was indeed reduced.
Marfan syndrome is named after Antoine Marfan, the French pediatrician who first described the condition in 1896. The gene linked to the disease was first identified by Hal Dietz and Francesco Ramirez in 1991.
Marfan syndrome (also called Marfan's syndrome) is a genetic disorder of the connective tissue. People with Marfan tend to be unusually tall, with long limbs and long, thin fingers.
The syndrome is inherited as a dominant trait, carried by the gene FBN1, which encodes the connective protein fibrillin-1. People have a pair of FBN1 genes. Because it is dominant, people who have inherited one affected FBN1 gene from either parent will have Marfan syndrome.
Marfan syndrome has a range of expressions, from mild to severe. The most serious complications are defects of the heart valves and aorta. It may also affect the lungs, the eyes, the dural sac surrounding the spinal cord, the skeleton and the hard palate.
In addition to being a connective protein that forms the structural support for tissues outside the cell, the normal fibrillin-1 protein binds to another protein, transforming growth factor beta (TGF-β). TGF-β has deleterious effects on vascular smooth muscle development and the integrity of the extracellular matrix. Researchers now believe, secondary to mutated fibrillin, excessive TGF-β at the lungs, heart valves, and aorta weakens the tissues and causes the features of Marfan syndrome. Since angiotensin II receptor antagonists (ARBs) also reduce TGF-β, ARBs (losartan, etc.) have been tested in a small sample of young, severely affected Marfan syndrome patients. In some patients, the growth of the aorta was indeed reduced.
Marfan syndrome is named after Antoine Marfan, the French pediatrician who first described the condition in 1896. The gene linked to the disease was first identified by Hal Dietz and Francesco Ramirez in 1991.